Avant 1963, synthétiser un peptide en laboratoire de chimie était une épreuve de patience. Chaque liaison peptidique à former exigeait des semaines de purification après chaque étape — cristallisation, recristallisation, chromatographie. Un peptide de 10 résidus prenait des mois. Un peptide de 30 résidus était impensable à l'échelle industrielle.
Le 22 mai 1963, Bruce Merrifield publie dans le *Journal of the American Chemical Society* un papier intitulé sobrement *« Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide »*. C'est l'une des innovations méthodologiques les plus transformatrices du XXe siècle en chimie organique. L'idée centrale : fixer le peptide en croissance sur un support solide (une bille de résine de polystyrène) pour pouvoir laver simplement les réactifs excédentaires à chaque étape, au lieu de purifier le produit intermédiaire entre chaque liaison.
Conséquence : un peptide de 10 résidus passe de plusieurs mois à quelques jours. La chimie peptidique devient industrialisable. Vingt-et-un ans plus tard, en 1984, Bruce Merrifield reçoit le Prix Nobel de chimie pour cette invention. Sans la SPPS (synthèse peptidique solide), ni le sémaglutide, ni le tirzepatide, ni l'octréotide, ni la quasi-totalité des peptides médicaments commercialisés aujourd'hui n'existeraient.
Au programme : l'histoire de l'invention par Merrifield, le mécanisme en quatre étapes répétées qui rend la méthode automatisable, les deux stratégies modernes (Fmoc et Boc), l'industrialisation actuelle et les limites de la méthode (peptides courts vs longs).
1963, Université Rockefeller — Merrifield publie la première SPPS
Le contexte de la découverte : Robert Bruce Merrifield, jeune chimiste de l'Université Rockefeller à New York, travaille depuis le milieu des années 1950 sur les peptides bioactifs (notamment la bradykinine, un peptide vasoactif de 9 acides aminés). Il se confronte directement au problème de productivité des méthodes en solution alors dominantes.
L'intuition centrale
L'idée de Merrifield est conceptuellement simple mais radicale : et si on attachait le peptide en croissance à un objet insoluble ? Les réactifs en excès (acides aminés activés, agents de couplage) resteraient en solution, le peptide attaché resterait sur l'objet. Une simple filtration suffirait alors à séparer le peptide des réactifs résiduels — au lieu d'une purification complète après chaque étape.
Le choix de la résine
Merrifield choisit comme support une résine de polystyrène chlorométhylé (résine de Merrifield). Les billes de résine sont gonflables dans les solvants organiques mais restent solides, faciles à filtrer. Un acide aminé peut être attaché à la résine par une liaison ester clivable plus tard.
La synthèse pivot
Le papier 1963 décrit la synthèse du tétrapeptide Leu-Ala-Gly-Val (un fragment de la bradykinine). Quatre cycles de couplage / déprotection. Démonstration de principe — la méthode marche, est reproductible, et offre une productivité radicalement supérieure aux méthodes en solution.
Réception initiale
Les premiers chimistes peptidiens accueillent l'innovation avec un mélange de scepticisme (la méthode produit-elle des peptides vraiment purs ?) et d'enthousiasme (la productivité est indéniable). Dans les années 1965-1975, l'automatisation des étapes répétitives devient possible — premiers synthétiseurs peptidiques automatisés commerciaux. Dans les années 1980, la méthode s'impose comme standard.
Prix Nobel de chimie 1984 — Merrifield seul, en reconnaissance d'une innovation méthodologique qui a démocratisé toute la chimie peptidique. C'est un rare exemple de Nobel attribué pour une méthode plutôt que pour une découverte fondamentale — soulignant l'importance des innovations instrumentales en science.
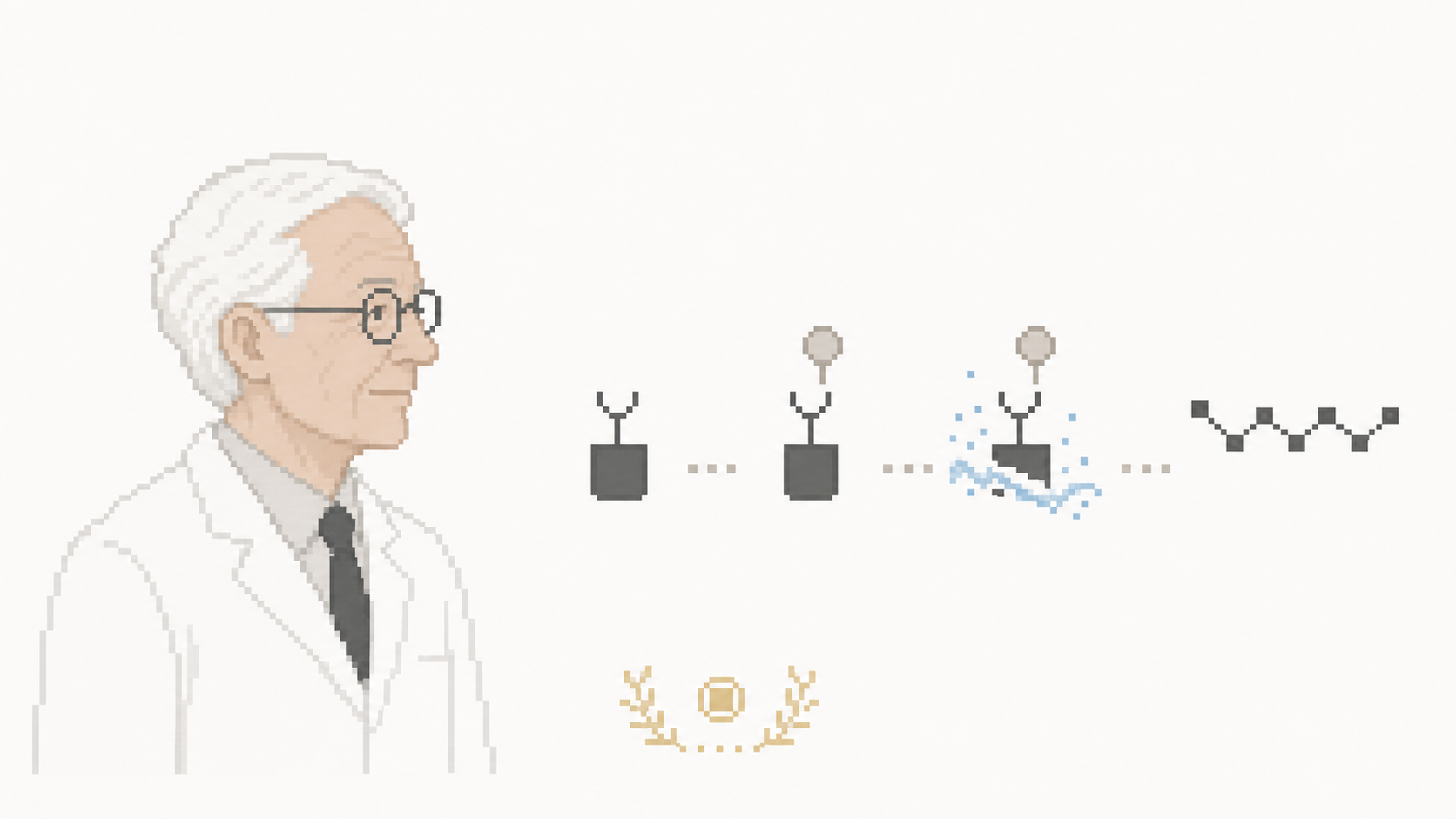
Le mécanisme en 4 étapes répétées
La SPPS suit un cycle conceptuellement simple, répété pour chaque acide aminé ajouté.
Préparation
Le premier acide aminé (typiquement le résidu C-terminal du peptide visé) est attaché à la résine par une liaison ester. Son groupe amine N-terminal est protégé par un groupe protecteur (Fmoc ou Boc, voir section suivante). Les chaînes latérales avec fonctions réactives (lysine, arginine, sérine, cystéine, tyrosine, etc.) sont également protégées par des groupes spécifiques.
Cycle de couplage (×N pour N résidus)
Étape 1 — Couplage : on ajoute l'acide aminé suivant, activé sur son groupe carboxyle (typiquement par un carbodiimide comme DIC ou DCC, parfois avec un additif comme HOBt ou OxymaPure). Le groupe carboxyle activé réagit avec le groupe amine libre du peptide en croissance attaché à la résine — formation d'une nouvelle liaison peptidique. Excès de 3-5 fois pour pousser la réaction à >99 % de rendement.
Étape 2 — Lavage : filtration de la résine et lavages successifs (DMF, méthanol, DCM) pour éliminer les réactifs en excès, les sous-produits, l'acide aminé activé non couplé.
Étape 3 — Déprotection N-terminale : retrait du groupe protecteur du nouvel acide aminé ajouté, pour libérer son amine et préparer le couplage suivant.
Étape 4 — Lavage à nouveau : élimination des produits de déprotection.
Le cycle est répété pour chaque résidu — un peptide de 30 résidus implique 30 cycles de couplage, soit environ 8 heures de synthétiseur automatisé en mode standard.
Clivage final
Une fois la séquence complète assemblée, le peptide est libéré de la résine par traitement chimique :
- Stratégie Fmoc : clivage par TFA (acide trifluoroacétique) à concentration élevée (~95 %), avec des *« scavengers »* pour piéger les cations protégés réactifs (eau, triisopropylsilane, dithiothréitol)
- Stratégie Boc : clivage par HF (acide fluorhydrique) liquide — méthode historique, conditions plus dures, équipements spécialisés requis
Le clivage retire simultanément les protections des chaînes latérales (généralement), libérant le peptide final brut. Purification ensuite par HPLC préparative pour atteindre la pureté pharmaceutique (typiquement > 95 % à 99 %).
Automatisation
Les étapes répétitives sont automatisables. Les synthétiseurs peptidiques automatisés commerciaux (Liberty Blue de CEM, Symphony de Protein Technologies, etc.) gèrent en continu les cycles de couplage, lavage et déprotection. Une synthèse de 30 résidus prend environ 8-12 heures sans intervention manuelle.
Stratégies Fmoc et Boc — deux groupes protecteurs N-terminaux
Le choix du groupe protecteur N-terminal est l'une des décisions structurantes en SPPS. Deux stratégies dominent.
Boc — la méthode historique de Merrifield
Boc = *tert-butyloxycarbonyle*. Méthode originale de Merrifield (1963).
- Protection : ajoutée sur l'amine N-terminale lors de l'achat de l'acide aminé
- Déprotection : par TFA dilué (50 % dans DCM) après chaque cycle
- Clivage final de la résine : par HF liquide anhydre — réaction très réactive, équipement HF dédié (matériaux résistants, capacités de neutralisation des résidus)
Avantages : robuste, validé pour des séquences complexes. Inconvénients : conditions acides répétées (TFA après chaque cycle), équipement HF spécialisé pour le clivage final.
Fmoc — le standard moderne
Fmoc = *9-fluorénylméthyloxycarbonyle*. Introduit par Louis Carpino en 1972 puis popularisé pour la SPPS par Atherton et Sheppard dans les années 1980 (livre de référence 1985).
- Protection : ajoutée sur l'amine N-terminale lors de l'achat de l'acide aminé Fmoc
- Déprotection : par pipéridine (20 % dans DMF) après chaque cycle — milieu basique doux
- Clivage final de la résine : par TFA concentré (~95 %) — pas besoin de HF, conditions plus accessibles
Avantages : conditions plus douces, pas de cumul d'acidité, automatisation simplifiée, compatibilité avec acides aminés modifiés sensibles aux conditions acides. Inconvénients : la pipéridine peut catalyser certaines réactions secondaires sur des séquences spécifiques.
Dominance Fmoc depuis les années 1980
La stratégie Fmoc est devenue le standard industriel depuis le milieu des années 1980. Les peptides médicaments commerciaux produits en SPPS aujourd'hui (sémaglutide, tirzepatide, octréotide, liraglutide) sont quasi tous synthétisés en Fmoc.
La stratégie Boc reste utilisée pour :
- Certains peptides cycliques avec ponts disulfure complexes
- Séquences avec acides aminés sensibles aux bases (rares)
- Académie et fournisseurs spécialisés en peptides très purs
Industrialisation — comment les peptides médicaments sont produits aujourd'hui
La SPPS est passée de méthode académique à technologie industrielle massive en quelques décennies. État du marché en 2026.
Échelles de production
- Recherche académique : milligramme à gramme par synthèse
- Préclinique / Phase 1-2 : gramme à kilogramme
- Production commerciale grand peptide médicament : kilogramme à tonne par an
À titre indicatif, le sémaglutide commercialisé à l'échelle mondiale en 2024-2026 est probablement produit à hauteur de plusieurs tonnes par an au total — toutes formulations confondues (Ozempic, Wegovy, Rybelsus). Le tirzepatide approche des niveaux similaires.
Sociétés spécialisées
Quelques acteurs majeurs de la production peptide industrielle :
- Bachem (Bubendorf, Suisse) — l'un des leaders mondiaux, fournisseur de nombreux peptides médicaments
- PolyPeptide Group (Strasbourg, France + sites internationaux) — acteur européen majeur
- Lonza (Suisse) — multi-produits pharmaceutiques incluant peptides
- Almac Group (Royaume-Uni) — services contractuels
- Pharmas internes : Novo Nordisk, Eli Lilly, Novartis ont aussi des capacités de production peptide internes
Ces sites opèrent sous Bonnes Pratiques de Fabrication (BPF (Bonnes Pratiques de Fabrication)/GMP), avec audits réguliers de l'EMA, FDA, ANSM, MHRA. La traçabilité lot, la pureté > 99 %, la stérilité sont garanties pour les peptides médicaments approuvés — c'est ce qui distingue radicalement la production pharmacie agréée du marché parallèle (étiquettes *« research only »* sans BPF).
L'exception insuline
L'insuline humaine recombinante (Humulin, 1982 par Eli Lilly) est produite par génie génétique chez *E. coli* (sous-unités A et B exprimées séparément puis assemblées) ou chez *Saccharomyces cerevisiae* (Novo Nordisk, expression directe de la pro-insuline avec maturation). Pour les peptides de cette taille (51 acides aminés en deux chaînes), la SPPS est techniquement possible mais économiquement défavorable — le génie génétique est plus efficace en termes de rendement et de coût.
Les autres peptides médicaments commerciaux (sémaglutide ~30 aa modifié, tirzepatide ~40 aa modifié, octréotide 8 aa cyclique, liraglutide ~30 aa modifié) sont produits par SPPS parce que leur structure (acides aminés modifiés, modifications post-synthétiques, chaînes d'acides gras attachées) n'est pas accessible directement par génie génétique.
La synthèse peptidique solide inventée par Bruce Merrifield en 1963 est l'innovation méthodologique qui a rendu possible la production industrielle des peptides médicaments. Sans elle, ni les analogues GLP-1 (sémaglutide, tirzepatide, liraglutide), ni les analogues somatostatine (octréotide), ni les analogues GnRH (leuproréline), ni les peptides cosmétiques (Argireline, Matrixyl, GHK-Cu), ni les peptides expérimentaux du biohacking (BPC-157, TB-500, CJC-1295, mélanotan-2) n'auraient pu exister à grande échelle.
C'est l'un des rares cas où le Prix Nobel récompense une méthode plutôt qu'une découverte fondamentale — illustration de l'importance des innovations instrumentales en science pharmaceutique. La SPPS reste aujourd'hui le standard industriel, automatisée par des synthétiseurs robotisés, opérée en BPF pharmaceutique, à l'échelle de tonnes par an pour les peptides les plus utilisés.
Pour comprendre les éléments construits par SPPS, voir acide aminé (les 20 briques) et liaison peptidique (la chimie qui les relie). Pour la définition complète du peptide et la frontière avec la protéine, voir le hub définition.
Sources
4- 01
- 02
- 03
- 04